











在韩国,医疗器械由食品药品安全部(MFDS)和保健福祉部(MOHW)监管。如果商家在韩国发布和销售的商品属于医疗器械,则应查阅关于医疗器械的《医疗器械法》、《医疗器械法实施细则》(《实施细则》)、《兽药等处理规则》、《医疗器械项目及各项目评级的规定》、以及《医疗器械标签及标签规定等》等法律法规,确保发布和销售的商品符合适用法律法规和MFDS、MOHW的要求。本文为商家提供有关韩国市场医疗器械资质的合规性参考,以帮助商家更好地履行医疗器械产品的安全与合规义务,保障消费者的合法权益。
一、 医疗器械的定义与分级
根据《医疗器械法》第二条定义,医疗器械是指单独或组合用于人或者动物的产品,包括仪器、机器、装置、材料、软件或者类似产品(药事法规定的药品、准药品以及残疾人福利法规定的残疾人辅助器具中的假肢、辅助器具除外)。医疗器械的使用目的应属于以下范围:
-
- 诊断、治疗、减轻、治疗或预防疾病;或
-
- 诊断、治疗、减轻或纠正伤害或残疾的产品;或
-
- 检查、更换、改变结构或功能;或
-
- 控制怀孕。
根据《医疗器械项目及各项目评级的规定》第二条,医疗器械中,独立制造、销售的医疗器械零部件,其主要功能是展示医疗器械性能、实现使用目的,对安全性、有效性有要求的,可以单独划归医疗器械项目。如果两种或两种以上的医疗器械组合并作为单独的医疗器械使用,则整个器械可归类为一种医疗器械。
《实施细则》附录一根据医疗器械的风险高低,将医疗器械分为四个等级,具体包括:
-
- 一级:潜在危害性很小的医疗器械(如眼科显微镜、听诊器等);
-
- 二级:潜在危害性较低的医疗器械(如MRI、脉搏血氧仪、消毒器、脑电图仪等);
-
- 三级:具有导致重症的潜在危害性的医疗器械(如避孕套、气体麻醉系统等);
-
- 四级:具有高度危害性的医疗器械(如植入式除颤器、冠状动脉支架等)。
其中,潜在危害性的判断标准包括:
-
- 与人体接触的时间;
-
- 侵入程度;
-
- 是否向患者输送药物;
-
- 是否对患者产生生物学影响。
关于医疗器械分级目录,以及医疗器械分级的程序详情,请参见《实施细则》附录一、《医疗器械项目及各项目评级的规定》及其附录一。
二、 合规要求
产品与制造相关资质
对韩国制造的医疗器械而言,根据《医疗器械法》第六条、《实施细则》第三条,医疗器械的制造商需取得MFDS颁发的制造业务经营许可证(申请表参见《实施细则》表一,许可证样式参见《实施细则》表二)。同时,取得制造许可证的人需根据医疗器械的风险高低,取得针对医疗器械产品品目或者品目类别对应的制造许可证(申请表参见《实施细则》表三、许可证样式参见《实施细则》表四)、制造证明书(申请表参见《实施细则》表五,证明书样式参见《实施细则》表六)或制造申报证(申请表参见《实施细则》表七,申报证样式参见《实施细则》表七之二)。
医疗器械产品具体所需资质可大致总结如下:
-
- 一级:制造申报证;
-
- 二级:部分产品需要制造证明书,部分产品需要制造许可证;
-
- 三级、四级:制造许可证。
资质审批流程可参考下图(摘自MFDS-医疗器械资料馆-许可信息-许可手续)。其中一级、二级医疗器械由韩国医疗器械安全情报院(NIDS)具体负责审批,三级、四级医疗器械则由MFDS具体负责审批:
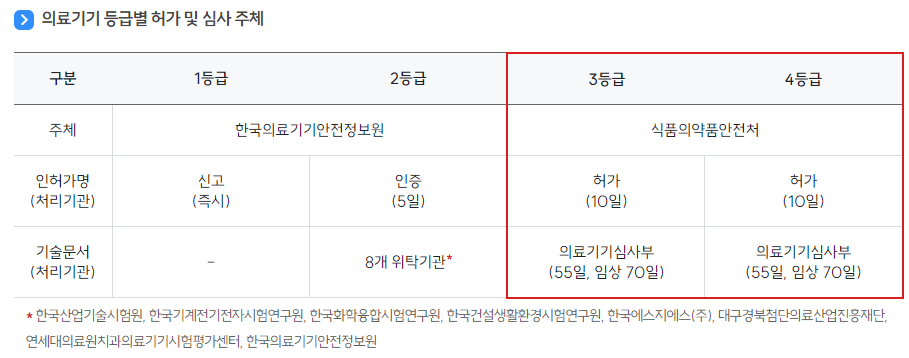
需要注意的是,医疗器械产品所需资质与医疗器械的分级并非完全对应,医疗器械产品具体需要取得制造许可证、制造证明书还是制造申报证,需按品目还是品目类别申请,需遵守《实施细则》第四条的规定。
进口相关资质
对进口医疗器械而言,根据《医疗器械法》第15条、《实施细则》第29条,欲从事医疗器械进口业务,必须取得MFDS颁发的进口业务经营许可证。取得进口业务经营许可的人对于拟进口的医疗器械,需根据医疗器械的风险高低,取得进口许可证、进口证明书或进口申报证。进口业务经营许可证、进口许可证、进口证明书、进口申报证的申请表和样式与上述产品与制造相关资质使用的申请表和样式一致。
同样,根据《实施细则》第34条,对于进口医疗器械产品具体需要取得何种资质的分类问题,准用《实施细则》第四条的规定,即与韩国国产医疗器械产品所需资质的分类方式一致。
销售相关资质
根据《医疗器械法》第17条、《实施细则》第37条、以及《兽药等处理规则》第23条,欲从事医疗器械销售业务,需要向管辖区的相关官员提交销售业务申报书(模板参见表36;兽用医疗器械参见表17)。受理之后,官员会向申请人出具销售业务申报证(模板参见表37;兽用医疗器械参见表18)。
但同时,《医疗器械法》第17条,以及《实施细则》第38条规定,销售如下所示的避孕用医疗器械,以及在医疗机构以外的场所使用的自我诊断用医疗器械时,可豁免销售业务的相关申报:
-
- 安全套;
-
- 具有或结合手机、家用电器等设备使用的血糖仪;以及
-
- MFDS根据产品风险和安全程度考量而通报的其他医疗器械,包括电子体温计、耳部红外线体温计、皮肤红外测温仪、自动电子血压计、用于自我诊断的移动医疗应及搭载该应用的产品(手机、平板电脑等)、颜色指示温度计,详见《医疗器械审批、报告、审查等规定》第55条。
关于申报文件中所填信息的具体要求,请参见《医疗器械审批、报告、审查等规定》第八至18条。
三、 安全标准
为了确保医疗器械的生产、进口和经营的安全性和适当性,韩国对医疗器械产品的适用范围、形状结构、检验规范、以及说明等都做了详尽要求。关于各类医疗器械的标准详情,请参见《医疗器械标准》及其附录一至三。
同时,根据《医疗器械法》第15条、《实施细则》第31条,取得进口业务经营许可证、进口许可证、进口证明书或进口申报证的医疗器械产品质量,必须符合相关生产设施及质量标准。具体标准详情参见《实施细则》附录四。
四、 标签及广告要求
韩国法规对医疗器械产品的包装和说明书做出了要求,进口、销售医疗器械的进口商和销售商必须严格遵守,以确保产品使用的安全性和适合性。根据《医疗器械法》第20条,产品容器或包装上需用韩文标示以下信息:
-
- 制造商、以及进口商的名称和地址
-
- 制造国
-
- 许可(证明或通知)号、名称(产品名称、项目名称、型号名称)
-
- 生产序列号和生产日期(如果有保质期,可以写保质期代替生产日期)
-
- 重量或包装单位
-
- 注明是“医疗器械”【의료기기】
-
- 如果是一次性的,请注明为“一次性”【일회용】和“禁止重复使用”【재사용 금지】
-
- MFDS与MOHW协商确定的医疗器械标准代码(指按照标准化体系(UDI)书写在容器或外部的数字、条形码(包括电子标签(RFID标签))等)
-
- 提供电子形式随附文件的网页地址
关于标签信息的具体格式要求,请参见《实施细则》第44条,以及《医疗器械标签及标签规定等》。关于产品随附文件所需陈列的事项要求,请参见《医疗器械法》第22条和《实施细则》第43条。
对于医疗器械的广告,MFDS也作出了相关规定。根据《医疗器械法》第24条、《实施细则》第45条,医疗器械的容器、外观、包装或者随附文件上不得标明或者书写与医疗器械有关的下列信息:①可能存在虚假或误解的事项;②未经核准或证明,或与申报事项不符的性能、功效或效果;③可能造成健康和卫生危害的使用方法或使用期限。关于医疗器械产品的广告可使用范围及限制的详细要求,请参见《实施细则》附录七。
五、 禁止销售拆封的无菌医疗器械
根据《医疗器械法》第18条之二、25条之五、以及《实施细则》第45条之四,任何人不得将法规规定的由生产商、进口商密封包装的医疗器械拆封后销售,这些医疗器械为①植入人体一年以上实行跟踪管理的无菌包装医疗器械,以及②在打开和分发时可能被污染或变质的下列医疗器械:
-
- 注射器(仅适用于无菌包装产品);
-
- 注射器针头(仅适用于无菌包装产品);
-
- 缝合器械;
-
- 伤口敷料(仅适用于无菌包装产品);
-
- 隐形眼镜;以及
-
- 其他经MFDS通报为无菌包装产品、配送时需密封的医疗器械。
六、 参考信息
【内容仅供参考,不构成任何法律意见或建议】
【结束】

相关导航



